The Hengduan Mountains (HDM) biodiversity hotspot of China exhibits exceptional plant diversity (about 12,800 seed plant species are recognized), with many different alpine plants present in the subnival zone.
One hypothesis for the rich biodiversity found in mountainous regions like the HDM is uplift-driven diversification; that is, orogeny produced topographical features creating diverse habitats and isolated populations, facilitating rapid intraspecific divergence leading to in situ speciation.
In addition, the connectivity of populations could have been influenced by climate fluctuations, contributing also to population divergence and speciation. However, the relatively few studies aimed at testing these hypotheses have employed a limited number of molecular markers.
In order to test this hypothesis, we investigate factors driving intraspecific divergence within a HDM alpine species Salix brachista (Cushion willow), a common component of subnival assemblages.
The research team produces a high-quality genome assembly for this species, with a genome size of ~340 Mb, contig N50 of 9.5 Mb and scaffold N50 0f 17.9 Mb, which is used as a reference for population genomics study of 77 individuals collected from 14 populations across the species’ distribution.
The team then characterizes genetic diversity, population structure and pattern of evolution by resequencing individuals collected across its distribution area.
Researches detect population divergence that has been shaped by a landscape of isolated sky island-like habitats displaying strong environmental heterogeneity across elevational gradients, combined with population size fluctuations have occurred since approximately the late Miocene.
These factors are likely important drivers of intraspecific divergence within Cushion willow and possibly other alpine plants with the similar distribution. Since intraspecific divergence is often the first step toward speciation, the same factors can be important contributors to the high alpine species diversity in the HDM.
Through comparative genomic analysis, The team found Cushion willow expanded and rapidly evolved gene familes significantly enriched in DNA repair and flavonoid biosynthesis, which might be involved in the adaptation of it to DNA damage induced by high level of UV radiation in high elevations.
The research detected distinct chromosomal rearrangement events in chromosomes 15 between Cushion willow and Salix purpurea. Chromosome 15 contains the sex-determination locus in Salix, which raises the possibility of the sex determination region undergoing evolution in the Salix lineage.
The result further indicated that high and lower elevation populations of Cushion willow face different types of natural selection, and detected some genes inferred to be important to adaptation to the different environmental conditions experienced at these sites.
This study was published in Nature communications with the title Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot.
The research was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDA 20050203.), Major Program of the NSFC (31590823), the National Key R & D Program of China (2017YF0505200), NSFC (31670198, 31560062, 31871271), the Science and Technology Research Program of KIB (KIB2016005), and the Youth Innovation Promotion Association, CAS.
The team of Prof. SUN Hang , Prof. YANG Yong-ping, and Dr. CHEN Jiahui at the Kunming Institute of Botany, Chinese Academy of Sciences and cooperators conducted this study.
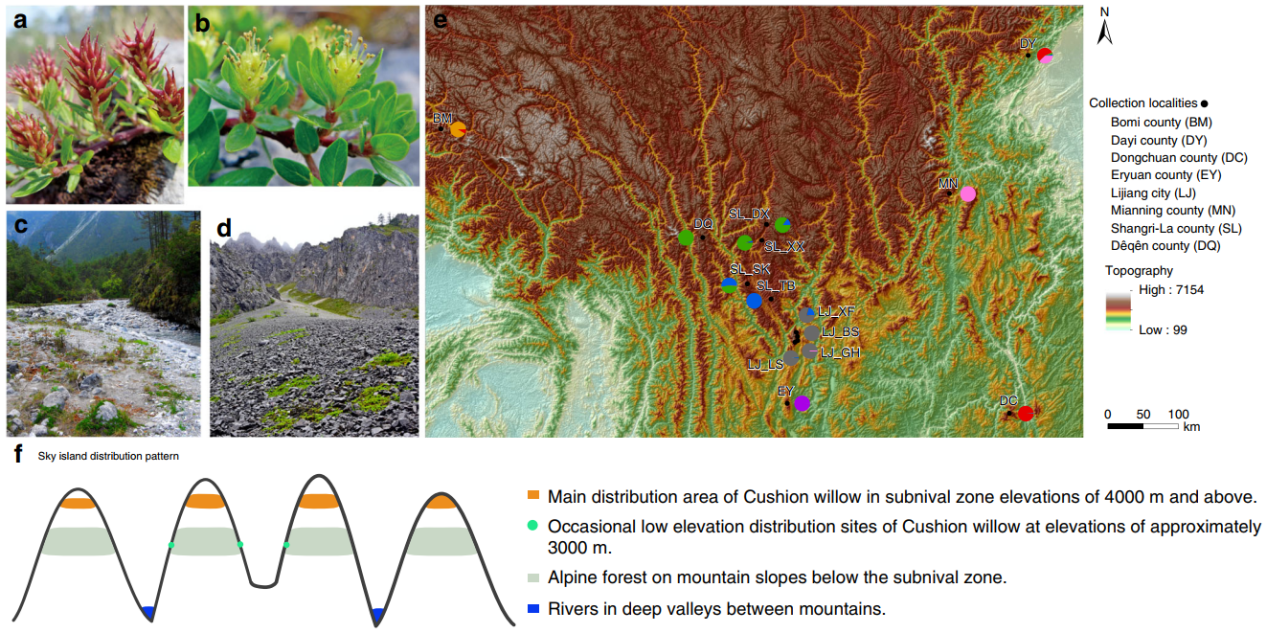
Figure 1. Cushion willow distribution pattern and locations of population samples (Image by KIB)
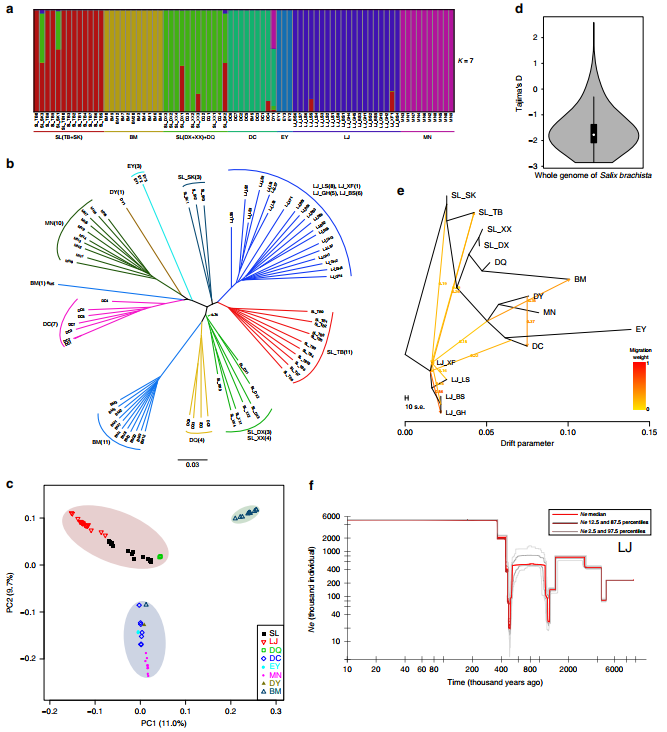
Figure 2. Population genetic strucure and demographic history (Image by KIB)
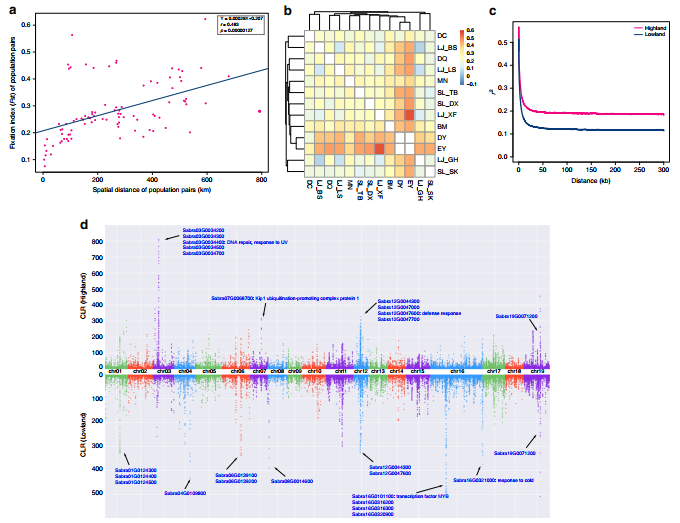
Figure 3. Signatures of selection in Cushion willow sampled from high and lower elevation sites (Image by KIB)
Contact:
YANG Mei
General Office
Kunming Institute of Botany, CAS
email: yangmei@mail.kib.ac.cn
(Editor:Yang Mei)



